
EPI-141 for RP4
Retinitis Pigmentosa (RP) is a group of genetic disorders characterized by degeneration of cells in the retina, which causes progressive vision loss, night blindness, and tunnel vision. RP is the leading cause of inherited blindness, with many different gene mutations involved in the disease, and an estimated prevalence of 1 in 4,000 people. One disorder in the RP umbrella is autosomal dominant retinitis pigmentosa 4 (adRP4), which is estimated to effect 5,000 to 8,000 people in the United States, with no available therapies.
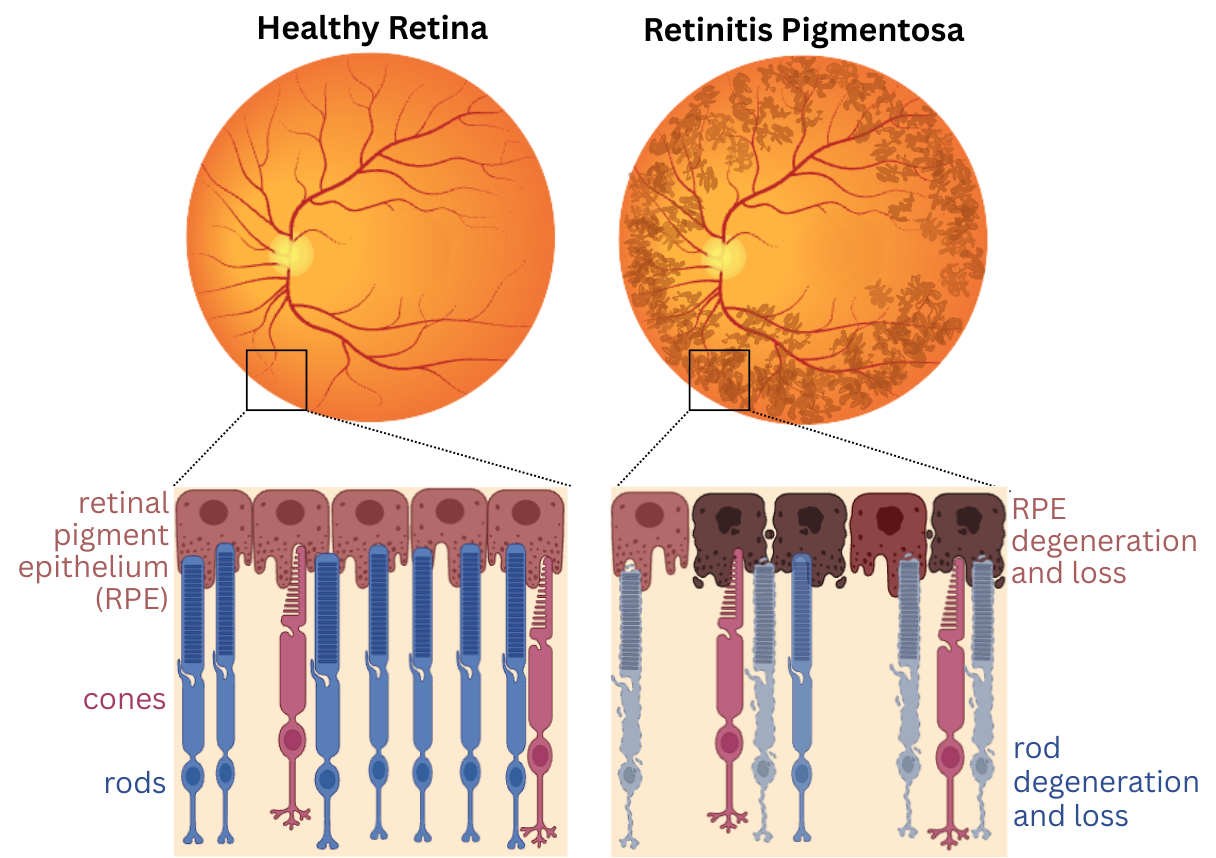
AdRP4 is caused by mutations in the Rhodopsin gene, which encodes a light-sensitive receptor protein involved in visual phototransduction. Rhodopsin mutations lead to accumulation of misfolded rhodopsin protein in the eye, which ultimately causes degeneration of light-sensing cells (photoreceptors). This condition is typically inherited in autosomal dominant manner, meaning that only one copy of a mutation in RHO is sufficient to cause adRP4. Over 200 different genetic mutations in the Rhodopsin gene have been identified as involved with adRP4, which has made developing gene therapy treatments for this disease challenging.
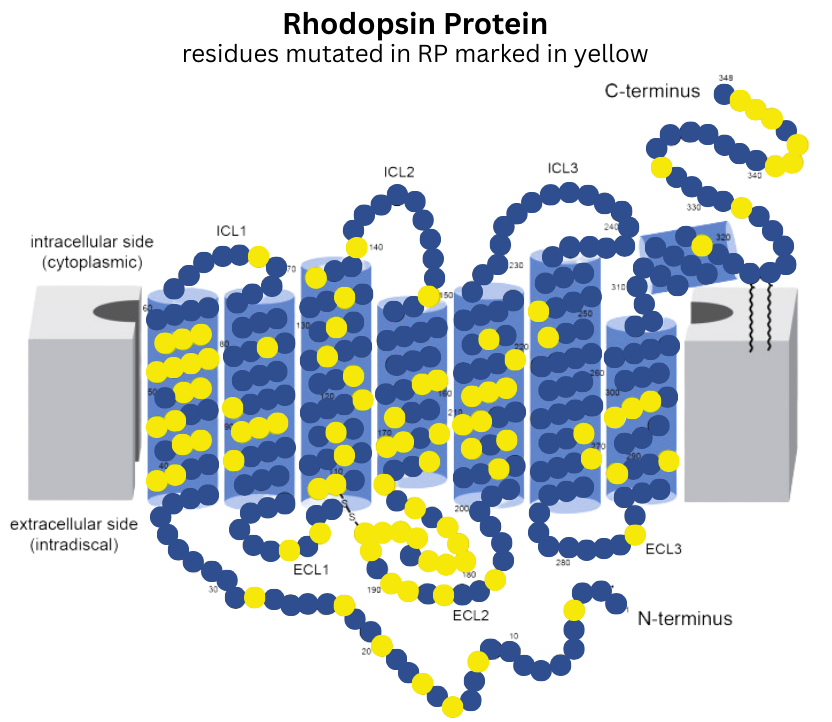
Image modified from PMID: 39791750
To meet this unmet patient need, Epicrispr is developing a unique treatment designed to work on any rhodopsin mutation without altering DNA directly.
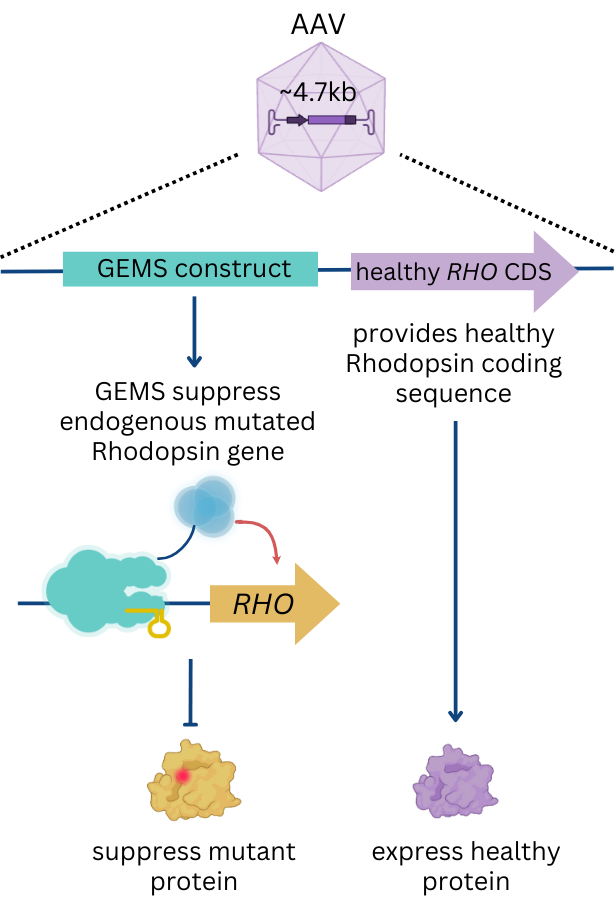
EPI-141 utilizes a “suppress and replace” strategy that simultaneously blocks mutated rhodopsin gene expression (suppress) while at the same time provides the healthy rhodopsin DNA sequence to produce a normal version of rhodopsin protein (replace), all delivered in a single AAV vector. Epicrispr’s technology and approach enables us to develop a single dose all-in-one mutation-agnostic treatment that can benefit the entire RP4 population.
Additional resources:
National Eye Institute: Retinitis Pigmentosa
American Academy of Ophthalmology: What is Retinitis Pigmentosa?